Automated model building with Buccaneer
From Media Wiki
Main Page - Using the CCP4 software - Model building with CCP4 - Automated model building with Buccaneer
Buccaneer is an automated protein model building program. It features robust handling of limited data resolution, and is competitive in terms of speed. It is particularly useful at resolutions of worse than 2.5A, although it can also be used at high resolution.
- Reference: K. Cowtan (2006) Acta Cryst. D62, 1002-1011. The Buccaneer software for automated model building
Contents |
[edit] Running Buccaneer
To run Buccaneer, you must first have a set of stucture factor magnitudes, and some estimated phases in the form of phase probability distributions, i.e. Hendrickson-Lattman coefficients or a phase and figure of merit. These will usually be obtained experimental phasing or from molecular replacement. You should run some sort of phase improvement before running buccaneer, see Phase improvement with CCP4.
Select the model building module from with CCP4i. There are two ways to run buccaneer: firstly as an automated model building and refinement cycle (Buccanner - build/refine), and secondly for model building alone (Buccaneer - fast build only). Select the 'Buccanner - build/refine' task.
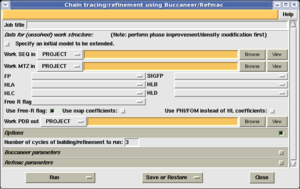
The buccaneer task interface looks like this:
In order to trun the program, you must provide 2 files:
- A sequence file. This must contain the sequence of the protein. The file may simply contain a list of 1-letter residue codes, or it may contain multiple chains, each specified by a chain ID (preceded with '>'), followed by the residue codes on subsequent lines (FASTA format).
If there is NCS present, then the NCS related chains need not be given (although the program will give the same results either way).
- An MTZ file. This must contain the stucture factor magnitudes, and phase probability distributions from after phase improvement. Normally these will be given as Hendrickson Lattman coefficients, although phase and figure-of-merit may also be used. (When rebuilding in a molecular replacement map, the phase and figure-of-merit may be obtained from the rigid-body-refinement.)
An output PDB filename is generated automatically. You may change this if you wish.
Select 'Run now' to start buccaneer.
[edit] Program output
The result of a Buccaneer is an atomic model, which is placed in the output PDB file. Use Coot or other model building software to view, correct, and refine the model. If necessary, buccaneer can be re-run to further extend the resulting model.
Some temporary files from the refinement steps are also available in the 'Show files from job' menu.
Double click your Buccaneer task in the CCP4i task list to see the log-file, which contains the output from successive runs of Buccaneer and Refmac. The logfile contains extensive diagnostic information from refmac. Select the 'View summary' button in the log file viewer to shows a brief summary, which reports how many chains and residues have been built in each cycle, and how many of those residues have been matched to the known sequence. If you know how many residues are expected in the assymetric unit of your structure, then the number of residues sequenced provides a good indication of the completeness of the model.
By default, the task performs three cycles of model building and refinement as follows:
| Cycle 1: | Run 3 cycles of buccaneer model building. Run 10 cycles of refmac refinement. |
|---|---|
| Cycle 2: | Run 1 cycle of buccaneer model building. Run 10 cycles of refmac refinement. |
| Cycle 3: | Run 1 cycle of buccaneer model building. Run 10 cycles of refmac refinement. |
The log file contents reflect this sequence.
[edit] Advanced options
[edit] File options
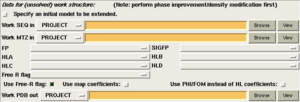
The following options affect the input files provided to the program:
- 'Specify an input model to be extended'. Check this box and specify an input PDB file if you wish to extend an existing atomic model instead of building a new model from scratch. Use this if you are trying to complete an MR model, or a model produced by Buccaneer and then corrected manually. The input model will on the whole be unmodified, but portions may be updated or even replaced by newly built chains.
- 'Use Free R-flag'. Uncheck this box if you do not have a Free-R set for your data. (Not recommended).
- 'Use map coefficients'. Check this box if you have map coefficients for a 'best' likelihood map into which you wish to build, for example FWT/PHWT columns from a refinement program.
- 'Use PHI/FOM instead of HL coefficients'. Check this box if you do not have Hendrickson Lattman coefficients. You must have a phase and figure-of-merit instead. If you have neither, then you need to perform either experiemental phasing and phase improvement, or molecular replacement and rigid body refinement, before running Bucccaneer.
[edit] Control options
The following option controls the iteration of the model building and refinement process.
- 'Number of cycles of building/refinement'. This option controls how many cycles of alternating model building and refinement will be performed. The default is 3. If after 3 cycles, the model is incomplete and sinificant new residues were built on the thrid cycles, it may be worth simply trying more cycles. Otherwise, some manual rebuilding may be required to provide a new model for Buccaneer to extend.
[edit] Model building options
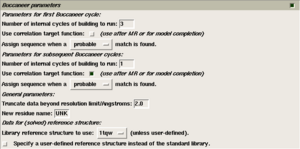
There are a number of parameters which can be changed to control the model building steps:
- Parameters for the first Buccaneer cycle.
These are the parameters which will be used by the first run of Buccaneer, before any refinement.- 'Number of internal cycles'. By default, the first run of Buccaneer performs 3 cycles of finding, growing and sequencing protein chains.
- 'Use correlation target function'. By default, the likelihood target is used for identifying protein features. This is best when starting from experiment phasing.
- 'Apply sequence when a --- match is found.'. By default, sequences are docked if the match is reasonably good. This can be changed to only sequence very good matches, or to sequence any plausible match.
- Parameters for the subsequent Buccaneer cycles.
These are the parameters which will be used for subsequent runs of Buccaneer, when it is being used to extend the refined model.- 'Number of internal cycles'. By default, subsequent runs of Buccaneer performs 1 cycle of finding, growing and sequencing protein chains.
- 'Use correlation target function'. By default, the correlation target is used for identifying protein features. This is best when extending an existing model.
- 'Apply sequence when a --- match is found.'. By default, sequences are docked if the match is reasonably good. This can be changed to only sequence very good matches, or to sequence any plausible match.
- General parameters.
- 'New residue name' is the name which is given to unsequenced residues. The default is 'UNK', change this to 'ALA' if you need to use the model in a program which doesn't recognise 'UNK'.
- 'Truncate data beyond resolution limit/Angstroms'. Use of high resolution data in Buccaneer makes the calculation slower and more memory-hungry, and does not contribute signficantly to the quality of the final model. Therefore, by default, data beyond 2.0A is truncated, unless you change this value.
- Data for (solved) reference structure.
This is the data which is used to calculate the likelihood targets which will be used to identify features in the unknown map. You should not need to change this.
[edit] Refinement options
The 'Refmac matrix weight' is used to control the relative weight of the X-ray and geomotry terms in refmac. Low values give more weight to the geometry terms, high values to the X-ray terms. The default value is 0.1. This is good for initial model building, especially at low resolution. At high resolution you may be able to increase this value to get a lower R-factor.
[edit] Related pages
It is also possible to use Buccaneer as a stand-alone program without Refmac. This gives access to a greater range of program options. See Fast model build with Buccaneer.
[edit] Program documentation
The latest version of the documentation is available here. This provides information on program keywords which may be used from the command line.
This page describes Buccaneer version 1.0.0 (CCP4 version 6.1.0).
--Kevin Cowtan 05:59, 18 April 2008 (CDT)