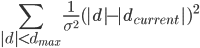
Please note that most of these keywords were in the previous versions also. Some of the keywords were implemented as user requests.
The numbers inside the brackets (if present) after the keyword indicates the version when this particular option became available.
LABIN FP=<label> SIGFP=<label> IP=<label> SIGIP=<label> F+= <label> SIGF+=<label> F-=<label> SIGF-=< label> HLA=<label> HLB=<label> HLC=<label> HLD=<label> PHIB=<label> FOM=<label> FREE=<label>
If only FP, SIGFP have been defined then a simple maximum likelihood refinement will be carried out
If F+, F-, SIGF+ and SIGF- have been defined then refinement using multivariate SAD function will be carried out.
If IP and SIGIP have been defined then refinement against intensities will be carried out. In the current version this option works only with twin keyword
If HLA, HLB, HLC, HLD have been defined then the external phase information will be used. These coefficients are usually generated by heavy atom refinement programs.
If PHIB and FOM have been defined then again external phase information will be used. Note that HLA, HLB, HLC and HLD contain more information about phases than PHIB and FOM
Note: In the current version external phase information will not work with twin, SAD options. They will work together in future versions.
If no labin keyword is given then the program will try to find amplitudes of experimental intensities and corresponding sigmas as well as FreeR_flag labels and carry out simple refinement. If no labin keywords and REFI SAD keyword has been defined then the program will try to find labels for Friedel pairs, corresponding sigmas and will carry out SAD refinement
Simple refinement: labin FP=FP SIGFP=SIGFP FREE=FreeR_flag
Refinement with the external experimental phases: labin FP=FP SIGFP=SIGFP HLA=HLA HLB=HLB HLC=HLC HLD=HLD FREE=FreeR_flag
Refinement with the SAD function: labin F+=F+ SIGF+=SIGF+ F-=F- SIGF-=SIGF- FREE=FreeR_flag
Using intensities: labin IP=I SIGIP=SIGI FREE=FreeR_flag twin
Current version takes only one keyword twin. All decisions are made automatically.
Note that in the current version of refmac (5.5.0031) twin refinement is not compatible with SAD or phased (using HLA, HLB, HLC and HLD) refinement. We are working on this
twin
This keyword gives a signal to the program. When Refmac sees this keyword it switches to twin refinement.
The program will find twin operators using tolerance level 0.001 and then using Rmerge values for each operator will make decision if the operator can be twin operator. After the first cycle of refinement it will remove all twin domains with fraction less than 5% making sure that the remaining operators together with the crystallographic ones form a group.
If twin keyword is defined then intensities can be used for refinement . See labin keyword above.
Other keywords
twin operator < operator > # It is not active yet
twin domain fraction < value > # It is not active yet
twin tolerance < value > # Active from the version 5.6.0051
This controls tolerance level that allowed symmetry constraints on cell parameters. Default value is 0.02. For merohedral cases this value is always 0, i.e. symmetry of lattice is higher. For pseudo merohedral cases higher tolerance may cause resolution stretching problem. In general if symmetry of lattice is very approximate then it is better to consider them as non-merohedral twinning cases. The reason is that in these cases in some directions overlap of reflections may overlap more than in other directions. Moreover at higher resolutions spots may become resolved.
twin FilterLevel < value > # Active from the version 5.6.040
Smallest allowed twin fraction. Default value is 0.05
twin rmergelevel <value> # Active from the version 5.6.0051
Larges Rmerge allowed. If Rmerge corresponding potential twin operator is larger than this value then this operator is discarded from further consideration. Default value is 0.5. If one wishes to check if reindexing is needed then this value could be increased to 0.6 or even 0.8. In these cases all potential twin operators will be accepted for further consideration and twin fractions will be calculated. If reindexing is needed then the first domain will have smaller estimated twin fraction than others.
The SAD target function performs refinement using the experimental phase information directly (using the SAD data and anomalous scatterers positions). To use SAD function appropriate labels (F+, F- and corresponding SIGF+, SIGF-) should be defined using labin keyword. Furthermore, at least one atom must have non-zero f''. The anomalous formfactors can be defined by the keywords:
anomalous formfactor [Name] [f'] [f'']
It will modify form factor of the given atom
anomalous wavelength [wavelengh]
If the wavelength is given then form factors (f' and f'' of all atoms) will be calculated using crossec. If for some element explicit formfactors are given then they will be used, for other atoms formfactors will be calculated. If wavelength is not given and mtz file has the wavelength then it will be used. If wavelength is not given and mtz does not have wavelength f'=0 and f''=0 will be used.
Refmac can also perform SAD phasing and refinement of substructure only. FB and PHIB output columns are generated for this case. No special input keywords are required, if Refmac sees substructure only in the pdb then it will switch. refi substructure keyword can be used to force the substructure phasing and refinement if needed from some reason.
Occupancies of all anomalous scatterers are refined by default if SAD target is used. Their refinement can be disabled by
refine orefine no
The SIRAS target function performs refinement using all native and derivative F+,F- data simultaneously. The FN, F+ and F- along with the SIGN, SIGF+ and SIGF- labels need to be defined by labin in order to use the target. Furthermore, at least one atom must have non-zero f'' and heavy atom substructure of the derivative compound needs to be specified by XYZIN2. Isomorphism between the native and derivative is assumed at the current implementation. Different models of the native and derivative and their simultaneous refinement with restraints between them is planned for the future.
Example:
weight auto | matrix [value]
If auto option has been given then the program will try to adjust weight parameter between X-ray and geometry. Current criterion is very simple: The program makes sure that rmsd bond from ideal values is between 0.015 and 0.025. If matrix value has been specified then it may be necessary to run the program several times and control rmsd for geometric parameters (e.g. bond lengths, angles)
mapc free | coefs | shar
free : Subkeyword that controls behaviour of free reflections for map calculation Possible values are: include - free reflections are included, exclude - free reflections are excluded or restore - free reflections and missing reflections are estimated (see below map coefficients ). Default value is restore
coef: Subkeyword for user defined map coefficient calculation. Values of the subkeyword are: n,m It will force the program to produce map coefficient nFo-mFc. mtz labesl for these coefficients are F_user, PHI_user. Normal 2fo-fc and fo-fc type map coefficients are always calculated.
shar: subkeyword for map sharpening. Value of this subkeyword is a bvalue that is used for all map coefficients. Output coefficients are modified using: Fcoef * exp(bvalue *|s|^2/4)
When calculating map coefficients REFMAC by default tries to restore missing reflections. Statistical basis of this is that expected value of unknown structure factors for missing reflections are better approximated using DFc than with 0 values. Of course to restore missing reflections accurately one needs full integration over all unknown parameters with their appropriate probability distributions that is not feasible in the current version. However approximate integration gives the value DFc. Current approach is trade off between bias introduced by restoring and noise level introduced by using zero values for missing reflections. Note that since for restored structure factors DFc is used and D reflects error in parameters, the level of bias is reduced substantially. If one wants not to include these reflections in the map calculation it can be done as follows: 1) Do not generate list of all unmeasured reflections; 2) use the instruction:
mapcalculate free exclude or mapcalculate free include
Another way of not restoring unobserved reflections is to use sigmas when calculating map. I.e. use only those reflection for map calculation for which sigma > 0.0. It can be done in fft or fftbig of ccp4 suite but not guaranteed to work in other software.
Unobserved reflections and their effect in map is a huge and underestimated problem that needs to be treated accurately.
Anomalous and difference anomalous maps can be generated. They are automatically generated if SAD refinement is performed.
If SAD refinement is not performed then the following keyword must be used to generate (weighted) coefficients for these maps:
There are two options for bulk solvent:
1) Babinet's bulk solvent. It is activated using
scale type bulk
keyword. The full keyword (or set of keywords) are:
scale type bulk
scale lsscalle fixbulk bvalue <value> scale <scale>
The first keywords signals the program that Babinet's bulk solvent should be used. It has effect that the total calculated structure factors are multiplied by the factor (1-kBulk exp(-BBulk |s|^2/4). kBulk and BBulk are in general adjustable parameters. The second keyword instructs the program to fix either kBulk or BBulk or both.
The second for of bulk solvent is mask based bulk solvent. It is activated (by default this form of bulk solvent is always calculated) using the following keyword:
solvent yes/no # Either use or not use mask based bulk solvent
solvent vdwprobe <value> ionprobe <value> rashrink <value>
solvent exclude DUM # Exclude atoms with name DUM from solvent mask calculation
These are parameters of the mask based bulk solvent. vdwprobe is a probe radius around vdw type of atoms, ionprobe is a probe radius around ion/polar/hydrogen bond capable atoms. rshrink is shrinkage radius after calculating mask with defined parameters. Default values are 1.2, 0.8,0.8
Algorithm for mask based bulk solvent
1) Increase radius of vdw atoms by vdwprobe and ion atoms by ionprobe.
2) Put zero inside the sphere centred at current atom with new radius
3) Reduce protein mask by rshrink. If points inside of the mask defined in the step 2) are closer than rshrink angstrom to the outside then define this point as being outside.
4) calculate structure factors and add them to the protein structure factors Fprotein+kmask exp(-Bmask |s|^2)/4) Fmask, where kmask and Bmask are adjustable parameters.
solvent optimise # This option is available from 5.6.0078
Optimise solvent parameters using grid search.
Keyword
make segid yes
Effect on other instructions:
Chain names involved in all instruction will be interpreted as segment id. For example NCS restraints could be:
ncsr nchains 4 chains AAss BAss TOss Yass nspans 1 1 100 1
The program will interpret AAss, BAss, TOss, Yass as segment ids. This instruction also affects records in the pdb header
External and user defined restraints
Current version of the program allows several types of external.
Distance restraints
external distance first chain [ch] residue [res] insertion [ins] - atom [n] [altecode [a]] second chain [ch] residue [res] insertion [ins]- atom [n] [altecode [a] ] value [v] sigma [s] [symm y/n] type [value]
external weight scale [value]
external weight gmwt [value]
external weight sgmn [value]
external weight sgmx [value]
external dmax [value]
This instruction will force to put restraints between defined atoms. Subkeywords insertion, altcode and symm are optional. If there is more than one restraint (including normal covalent bond restraint) then only the last one will be used.
Examples:
If one wants to restrict restraints to certain type of atoms then following command could be used
external use M|H|A
M - use main chain atoms only
H - use only those atom pairs that can make hydrogen boding pairs. Note that distance between these atoms are not analysed
A - or without thos keyword: use all atom pairs specified in the external restraints instructions
1) Restraint between atoms in the same asymmetric unit (without symmetry)
2) Restraint between atoms symmetry related atoms
exte dist first chain A resi 2 atom CA seco chain A resi 5 atom CA valu 4.0 sigm 0.02 symm Y
In this case all symmetry operators will be tried and that that brings these two atoms to the closest contact will be used for the restraint.
Angle restraints
external angle first chain [ch] residue [res] insertion [ins] -
atom [n] [altecode [a]] next chain [ch] residue [res] insertion [ins] atom [n] [altecode [a] ] [symm y/n] next chain [ch] residue [res] insertion [ins]-
atom [n] [altecode [a] ] [symm y/n] value [v] sigma [s] [symm y/n]
The three atoms are defined and the angle formed between these three atoms is restrained to the value defined by value with the sigma defined by sigma subkeyword.
external torsion first chain [ch] residue [res] insertion [ins] atom [n] [altecode [a]] next chain [ch] residue [res] insertion [ins] atom [n] [altecode [a] ] [symm y/n] next chain [ch] residue [res] insertion [ins] atom [n] [altecode [a] ] next chain [ch] residue [res] insertion [ins] atom [n] [altecode [a] ]
[symm y/n] value <v> sigma <s> period> <p>
External restraints could be saved in a file and used in refinement as:
refmac [all usual things] << eof
@file_external_restraints
all other instructions
eof
Harmonic restraints and special positions
Under the pressure from various users I have added harmonic restraints. If you use these restraints then atoms will be restrainted to their current position and movement from those positions will be slower than for other atoms. Syntax for restraints to keep atoms in special position is similar to harmonic restraints keywords. Keywords for harmonic restraints:
external harmonic|special chain [ch] residue [res] insertion [ins] atom [n] [altcode [a]] [sigma [value]]
or
external harmonic|special residues from [residue_number] [chain_name] to [residue_number] [chain_name] [atom <atname>] sigma [value] sigma 0.1
or
external harmonic|special atinfo chain [chain] residue <residue> atom <atom> sigma <value>
For example:
will put harmonic restraints on all of the atoms of the residues between 225A to 250A. The weight will be calculated using 1.o/sigma**2
external harmonic residues from 225 A to 250 A atom CA sigma 0.02
will put harmonic restraints on CA atoms of the residues from 225A to 250A.
external special atinfo chain A residue 255 atom CU
will keep CU in special position if its current coordinates are very close to special position (i.e. if difference between symmetric coordinates is less than 0.5A). Necessary symmetries are calculated on fly.
Harmonic distance restraints (Ridge regression)
Keywords:
ridge distance include all/none # default none
ridge distamce within chain <chain> resides <res1> <res2> sigma <sigma> dmax <dmax> # defaults for sigma and dmax are as define in the following kyewords
ridge distance sigma <value> # Default 0.02
ridge distance dmax <value> # Default 4.2
ridge distance hydrogens Y/N # use hydrogens. Default is N - do not use hydrogens in jelly body restraints
ridge atoms <sigma>
ridge bvalue <sigma>
If ridge distance sigma (default 0.02A) instruction has been given then the program will add the following function to the target function:
Where d is distance between atoms dcurrent is current distance between atoms. The program updates at every cycle dcurrent to the current distance between atoms.
If this instruction is defined then the program will calculate the list of all pairs of atoms for which distance between is less than dmax. Default value is 4.2.
Note that harmonic distance restraints will be applied together with non-bonded antibumping restraints.
If ridge atoms instructions is defined then the program adds the following function (it is same as harmonic restraitns applied to all atoms)
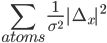
Where Δx is the shifts to be applied to the atomic positions
If ridge bvalue instruction is defined then the program adds the following term:
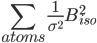
If ridge include all is not defined and instructions for particular regions are defined (e.g.
ridge distance within chain A residues 1 5
)
then restraints will be applied only to those atoms that belong to these regions . If ridge include all is defined then atoms outside user defined regions will also be restrained using default parameters (sigma and dmax).
Torsion angle restraints(from dictionary)
restr tors include | exclude
Include or exclude given torsion angle in the restraint calculation. Both subkeywords have the following syntax
resi | group | link [name] name [name] value [value] sigm [value] period [value]
For example
restr tors include resi VAL name chi1 value 60 sigma 2.0 period 3
This instruction will force chi1 torsion angle of all residues VAL to be restrained to 60 with period three.
Similarly this instruction can be applied to a group of residues (e.g. peptide, pyranose, DNA/RNA) or links between monomers (e.g. TRANS, ALPHA1-3 links). This restraint will be applied to all residues with name PHE.
An example how to exclude some torsion angles from restraints
restr torsion exclude residue PRO name chi1
If instruction is given up to the name of the monomer then all torsion angles in this monomer that have name starting with "var" will be restrained. For example:
restraint torsion include residue BLA
These instructions should be used with care. One should make sure that values used (in dictionary or defined by user in instructions) are valid and make chemical sense.
refinement exclude all from [residue] [chain] to [residue] [chain]
All atoms between given residues will be excluded from refinement (restraints, structure factor and gradient calculations), but they will be used for mask calculation.
Old instructions (for backward compatibility). One instruction per ncs group should be given:
ncsr nchains [nchains] chains [chain1] ... [chain_nchains] .. nspans [n1] [n11] [n12] .. [nn11] [nn12] [n4]
nchains - number of chains involved in this ncs chain - chains involved in this ncs n1 - number of spans n11,n12 - Start and end for the current ncs span
n4 - weighting options
New instructions (a little bit more flexible and useful for complex molecules):
Definition of ncs groups:
id - ncs id. It is used to group ncs related chains together. nchain - number of chains involved in this ncs. It defines the number of ncs matrices need to be calculated. residue - defines ncs restraint spans sigx - sigma on positional parameters sigb - sigma on atomic displacement parameters
Each ncsr id can have only one sigma (sigx) on positional and one on ADPS (sigb)
Example:
ncsr group 1 nchains 3 chains A B C residue_range 1 100 residue_range 201 300 residue_range 401 500
ncsr group 1 nchains 3 chains D E F residue_range 10 50
ncsr group 1 sigx 0.02
ncsr group 1 sigb 1.0
If you do not define rigid groups then the program takes each chain (if available then segment) as a rigid group. I.e. if you want to refine each chain (segment) as a separate rigid group then the following keyword is sufficient. Note that even if you do not use segment but they are defined in the input PDB then these segments will be used as rigid groups.
mode rigid
If you do not define TLS groups then the program takes each chain (if available then segment) will be as a TLS group. I.e. if you want to refine each chain (segment) as a separate TLS group then the following keyword is sufficient. Note that even if you do not use segment but they are defined in the input PDB then these segments will be used as rigid groups.
refi tlsc
If the keyword
tlsout addu
has been specified then the output file will contain ANISOU card for atoms involved in TLS group definitions. The values for ANISOU are contribution from TLS with added residual B values. In this case B value
Starting from the version 5.7.0018 refmac can be terminated during its run. It can either be terminated unconditionally or if one of the conditions is fullfilled: Rfactor is too higher, Rfree is too high, Rfactor jump is too high, Rfree jump is too high, differences between R and Rfree is too high.
Instructions determining termination are:
kill [file_name] # It is a signal that the program can be terminated and termination signals are in the file - file_name
File may contain one of the following instructions:
stop Y|1|T # Unconditional termination
stop N|0|F # contiditional termina
rfactor value # if Rfactor > value then terminate
rfree value # if Rfree > value then terminate
delta rfactor value # if rfactor jump > value then terminate
delta rfree value # if rfree jump > value then terminate
delta rrfree value # if Rfree-R > value then terminate
For full description of libcheck see Alexei Vagin's libcheck page
For smile string formats and syntax see: daylight site
To create a dictionary entry from SMILE string you need to have a file that contains SMILE for your ligand. One file should contain one ligand only. Then a dictionary entry can be created using libcheck:
libcheck file_smile [file] mon [give a reasonable name]
Then libcheck will create a dictionary entry. There should be one carriage return after the line libcheck and after the last instruction for the libcheck.
For mol2 see the mol2 manual and for sdf file see sdf manual
To create a dictionary entry from SYBIL MOL2 or SDF MOL file you need to have a file that contains ligand in one of these formats. Note that only 3D version of these formats can be used in this context. 2D version of these formats is equivalent to SMILE. That is why using SMILE strings in these cases seems to be more reasonable. Once you have file you can use libcheck to create a dictionary entry:
libcheck file_mol [MOL2 or SDF files] mon [mon name. It is optional]
The current version of libcheck creates dictionary from sdf v2000. V3000 has not been tested yet. If somebody wants to test please let me know.
In this sections protocols will be described using keywords. If you are using ccp4i then either there are appropriate options on the interface or you can create a file containing necessary keywords and then use "Developers option" to add this file. Then the keywords defined in this file will override the options defined in the ccp4i
Main ccp4 wiki. A lot of useful info. It is dynamic and is becoming a powerful resource
Dundee prodrg server It can create dictionary for ligands for refinement in refmac.
EBI MSD-CHEM server You can search for ligand you are interested in. Then save the results in cif format. This file can be used in libcheck to create complete description of the ligand.
Drugbank is another server that can be used to get "ideal" structures.
If you use REFMAC please cite to one of these papers!!!