Phase improvement by NCS averaging with DM
From Media Wiki
Main Page - Using the CCP4 software - Phase improvement with CCP4 - Phase improvement with DM
DM is a program which takes an initial set of phase probability distributions from experiment phasing (or occasionally molecular replacement), and produces an improved set of phase probability distributions which may be used to calculate a clearer and more interpretable map. It does this by applying real space constraints based on known features of a protein electron density map.
When Non-crystallographic symmetry (NCS) is present in the crystal, the following modifications are applied to the electron density:
- Solvent flattening. This technique reduces the noise in the disordered solvent region of the unit cell. The method implemented in DM uses a combination of solvent flattening and gamma correction to acheive the effect of solvent flipping.
- Histogram matching. This technique sharpens peaks in the protein region of the unit cell. It is particularly useful for imporving the resolution of the map.
- NCS averaging. This technique averages density between NCS related copies of the molecule, reducing the noise and increasing the constraints on the phases.
Contents |
[edit] Running DM for NCS averaging
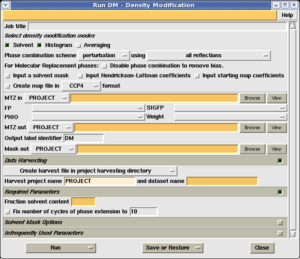
DM performs phase improvement to improve the phase estimates (or phase probability distributions) obtained from experimental phasing or molecular replacement, and thereby improve the electron density map. To run DM for NCS averagingm, you need three pieces of information: An MTZ file containing a set of observed structure factors and phase information, an estimate of the solvent content of the crystal, and some estimates of the NCS operators, expressed in terms of a rotation and translation.
Running the program, and setting the input MTZ file and solvent content are covered in Phase improvement with DM, and the steps on that page should be follwed first. For NCS averaging to be performed, there must be NCS present in the crystal, and the NCS operators must be determined.
[edit] Determining if NCS is present in the crystal
The following features may be indicators that non-crystallographic symmetry is present in the crystal:
- The cell content analysis (see Phase improvement with DM) shows that a single molecule is much smaller than the assymetric unit of the crystal, leading to an implausibly high solvent content.
- The self-rotation function (which may be calculated using MOLREP) shows a strong peak which does not correspond to a crystallographic symmetry axis.
- The patterson shows a strong peak which does not correspond to a lattice centering operator. This is suggestive of a pure translational NCS.
[edit] Determining the NCS operators
There are three main ways of determining the non-crystallographic symmetry operators: Using a partial model, using known heavy atom positions, or using density molecular replacement.
[edit] Determining the NCS operators using a partial model
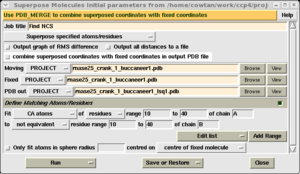
If a partial model is available, either from Molecular replacement, or from model building in an initial electron density map, this may be used to determine the NCS operators using the 'Superpose molecules' task from the 'Coordinate utilites' module.
From the top menu, select the 'Superpose specified atoms/residues' option. This allows specific residues to be chosen for superposition, and will output the result in the required format for DM.
Next, select your PDB file as both the moving and fixed molecule.
In the 'Define matching atoms/residues' folder, enter start and end residue numbers and a chain name to select the residue range of your first NCS copy. Then change 'equivalent' to 'not equivalent' in the next pull down, and enter the corresponding residue range and chain name for the second NCS copy. Select 'Run now'.

In the log file find the Euler angles, and the translation in orthogonal Angstroms, as shown on the right. These provide the information required for DM.
If you have more than two NCS related molecules, repeat the previous steps to get the operator matching the first molecule onto each of the others. For example, if you have 4 NCS related chains, A, B, C and D, then you need 3 operators: A->B, A->C, and A->D.
[edit] Determining the NCS operators using known heavy atom positions
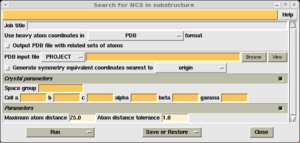
If heavy atom positions are availabe from experimental phasing (or occasionally from a difference map calculation), these may be used to determine the NCS operators using the 'Find NCS from heavy atoms' task from the 'Density improvement' module.
Select the PDB file containing your heavy atom coodinates. As long as this file contains the crystal information, no other input is required. Select 'Run now'.

The output from this task includes a list of similar triangles found amongst the heavy atoms, followed by a list of the corresponding NCS operators under the title "These will now be sorted according to Natoms and rotation loop" (as shown on the right). Following is a list of possible NCS operators in a variety of formats, also with some evaluation criteria:
- Natoms is the number of atoms related by this operator: if there a small number of operators for which this value is substantially higher than the rest, then these operators are probably right.
- Rotation order indicates if the operator forms a proper rotation. If this value is greater than 1, then it is the order of the NCS axis. If it is 0, then the operator is improper, which is less common but may still occur.
You should compare the number of likely operators against the expected number of molecules in the assymetric unit, as previously determined by the cell content analysis task.
[edit] Determining the NCS operators using molecular replacement
This is a complex task for experts only, but the steps are as follows:
- A single molecule or symmetric oligomer must be identified by visal inspection of the map.
- A mask must be drawn covering the identified region.
- The mask is used to 'cut out' the density of the identified region.
- This density is used as a molecular replacement search model in a molecular replacement program.
[edit] Entering the NCS operators
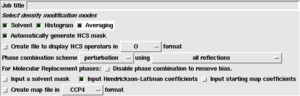
Select the 'Averaging' checkbox at the top of the task window (as shown on the right). A new folder will appear in the task window, labelled 'Define NCS Symmetry'.
Next you must add the NCS operators determined by one of the methods in the previous section. The first operator is always the identity operator - this is so that the number of operators matches the number of molecules.
To add the other operators, use the 'Add operator' button. Do not use the 'Add domain' button - that serves a different purpose. A new row of boxes appears where you may enter the 3 Euler angles and 3 orthogonal translation values. (As a convenient shortcut, you can select several numbers at once from another program or your notes, and paste them into consecutive fields using shift+right-mouse-button.
Once you have entered all the operators, select 'Run now'.
[edit] Program output


It is very easy to make a mistake when entering the NCS operators, so you should check the DM log file particularly carefully. Double-click your DM task in the CCP4i task window to view the log file. This contains two very good checks of whether the NCS operators are right:
- If the averaging mask is being calculated automatically, then has a sensible mask been produced?
- Are the correlations between the NCS related regions reasonable?
Both of these can be answered from the log file. The averaging mask determination can be assessed both on the basis of the mask statistics and the mask shape.
To check the mask statistics (right), find the word 'peaks' in the log file. You will find 4 consecutive peak lists like the one on the right. Scroll down to the fourth of these. If, as in this case, the mask consists of one large connected region and a handful of tiny regions, then it has worked. If the second peak is more than 20% of the size of the first peak, then there may be a problem.
The next section gives further statistics on the mask, followed by a representation of a mask section (right). This should usually be a single, compact blob.
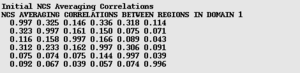
Finally, find the averaging correlation matrix (right). This matrix gives the corrlations between every pair of NCS related domains. In the case of Proper NCS only the top line is meaningful. The numbers on the top line give the agreement between the first copy of the molecule and each other copy. In this case, there are 6 copies (chains A-F). Chain A of course matches itself, so the first number is close to 1. Chain A agrees with chain B with a correlation of 0.326, wheras the correlation with chain C is only 0.146. Values of greater than 0.2 are generally correct, values less than 0.1 generally indicate an error with the operators or mask. In between, it is best to review the resulting map.
In the example on the right, the 3rd and 6th operators require further scrutiny.
[edit] Advanced options
[edit] General NCS Options
In order to generate an averaging mask covering the correct volume, DM must estimate the number of molecules related by Proper NCS. This is normally done automatically, however in complex cases of mixed crystallographic and non-crystallographic symmetry it may get it wrog. If in doubt, you can specify the number of monomers to mask here.
Additionally, in the case where an NCS axis is parallel to a crystallographic axis, the mask may be unbounded along one direction. This can be dealt with by specifying bounds for the mask.
[edit] Multi-domain averaging
If the molecule is flexible, then it is possible for the crystal to form in such a way that the different copies of the molecule in the assymentric unit are in different conformations. In this case it is necessary to divide the molecule into rigid domains, and average each domain with the other copies of that domain separately. The NCS operators will differ for each domain to account for the different orientations of each part of the molecule, although it may be possible to start with the same operators for each domain and allow the program to refine the differences.
To perform a multi-crystal averaging calculation, use the 'Add domain' button to add additional domains. You will then need to enter a full set of averaging operators for each domain.
Automatic mask calculation is not available for the multi-domain case, so you must generate a mask for each domain externally and supply this to DM.
[edit] Other options
The remaining options are as for DM.
[edit] Related pages
Phase improvement with DM for calculations without NCS.
[edit] Program documentation
The latest version of the documentation is available here. This provides information on program keywords which may be used from the command line.
This page describes DM version 2.1 (CCP4 version 6.1.0).
--Kevin Cowtan 06:01, 18 April 2008 (CDT)