Phase improvement with DM
From Media Wiki
Main Page - Using the CCP4 software - Phase improvement with CCP4 - Phase improvement with DM
DM is a program which takes an initial set of phase probability distributions from experiment phasing (or occasionally molecular replacement), and produces an improved set of phase probability distributions which may be used to calculate a clearer and more interpretable map. It does this by applying real space constraints based on known features of a protein electron density map.
In the simplest case, the following modifications are applied to the electron density:
- Solvent flattening. This technique reduces the noise in the disordered solvent region of the unit cell. The method implemented in DM uses a combination of solvent flattening and gamma correction to acheive the effect of solvent flipping.
- Histogram matching. This technique sharpens peaks in the protein region of the unit cell. It is particularly useful for imporving the resolution of the map.
Contents |
[edit] Running DM
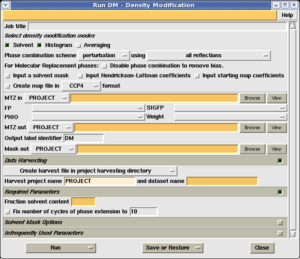
DM performs phase improvement to improve the phase estimates (or phase probability distributions) obtained from experimental phasing or molecular replacement, and thereby improve the electron density map. To run DM, you need two pieces of information: An MTZ file containing a set of observed structure factors and phase information, and an estimate of the solvent content of the crystal.
The easiest way to obtain an estimate of the solvent content is using the Matthews coefficient estimation. This is implemented in the 'Cell content analysis' task in the Density Improvement module.
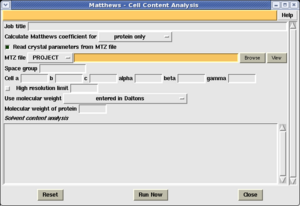
In order to estimate the solvent content, launch the 'Cell content analysis' task in the Density Improvement module. You must provide the following information:
- The MTZ file containing your observed structure factors.
- An estimate of the cell contents. You can provide this either in terms of the Molecular weight of the protein, the number of residues, or a sequence file, by making the appropriate selection from the 'Use molecular weight' menu.
Select 'Run now', and the program will produce a table of possible unit cell contents in the box at the bottom of the window.

An example of the output is shown on the right. In this case the program has produced 3 possible estimates of the solvent content, depending on whether there are 1, 2 or 3 molecules in the assymetric unit. Alongside each estimate is a likelihood of the value - this is a useful guideline, but no more.
In the case shown, the best estimate of the solvent content is 45%, based on 2 molecules in the assymetric unit.
Having obtained this estimate, launch the 'DM' task in the Density Improvement module. You can now enter the following information:
- The name of your MTZ file containing a set of observed structure factors and phase information. The phase information may be represented by a Best phase and Figure of merit, or by Hendrickson Lattman coefficients. Select the appropriate columns generated by the phasing program.
- The solvent content from the previous step, as a fraction of the total unit cell. So for example, in this case the solvent content is 45%, so the value 0.45 is entered in the box labelled 'Solvent content'.
The remainder of the fields should be filled out automatically. Select 'Run now'. The calculation should take less than a minute for all but the largest problems.
[edit] Program output
DM produces an extensive log file and graphs, including some complex analysis of the input structure factors. However, while this may be useful for spotting problems with the data, it does not generally give a good indication of how well the program has worked. The best ways to determine whether the resulting map is interpretable is to:
- Look at it in a graphics program, such as Coot.
[edit] Advanced options
[edit] Program mode
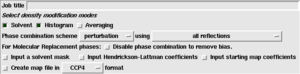
There are various other options which affect the calculation:
- Major options:
- Averaging: If there is non-crystallographic symmetry in the crystal, 'dm' can exploit this to obtain much better phasing. See Phase improvement by NCS averaging with DM.
- Minor options:
- Phase combination mode: Only change this if the program fails.
- Disable phase combination: This will reduce bias to an MR model, but will also lead to poorer maps unless NCS is available.
- Input a solvent mask: This overrides the internal automatic mask calculation.
- Input a Hendrickson Lattman coefficients: Allows updated HL coefficents to be output.
- Input starting map coefficients: Useful after MR if there are missing data.
- Create map file. Redundent, because modern graphics software will generate a map on the fly.
[edit] File options
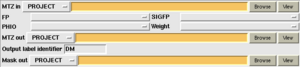
The following options affect the input files provided to the program:
- FP/SIGFP: Select the observed data (or recentred F's from a phasing program).
- PHIO/FOMO: Select the Best phase and Figure of merit from the phasing calculation.
The program produces an output MTZ file containing the improved phases, and a mask file (in CCP4 map/mask format) which can be viewed in a graphics program.
[edit] Other options
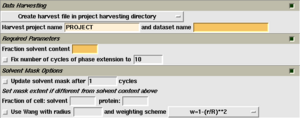
The following options affect the other features of the calculation:
- Fix number of cycles to: By default, DM runs until the map no longer seems to be improving. However, when averaging is present, it can often be beneficial to force the program to run more cycles: 10, 20 or even 100.
- Fraction of cell/solvent/protein: Occasionally solvent flattening can eliminate flexible loops from the protein. In this case it may be desirable to exercise finer control over the solvent mask. Changing the solvent content is inadvisable, since this affects the data scaling. Instead, these boxes can be used to control the fraction of the cell marks as solvent, protein, or neither. For example, the values solvent 0.30, protein 0.60 will lead to 30% of the cell being marked as solvent, 60% as protein, and 10% as unknown.
[edit] Related pages
If there is non-crystallographic symmetry in the crystal, 'dm' can exploit this to obtain much better phasing. See Phase improvement by NCS averaging with DM.
[edit] Program documentation
The latest version of the documentation is available here. This provides information on program keywords which may be used from the command line.
This page describes DM version 2.1 (CCP4 version 6.1.0).
--Kevin Cowtan 06:00, 18 April 2008 (CDT)