[Date Prev][Date Next][Thread Prev][Thread Next][Date Index][Thread Index]
RE: [ccp4bb]: planarity restraint in refmac5
I want to expand on Bart's comment. While it is entirely correct as he
states that if things go bad for reasons *recognizable* in (good) maps,
*unjustified* diversions from geometry make no sense, in particular if
preventing so lowers Rfree. I am just checking some log differences
between refmac4 and refmac5 w.r.t. the number of Xray vs geometric
restrains when using defaults.
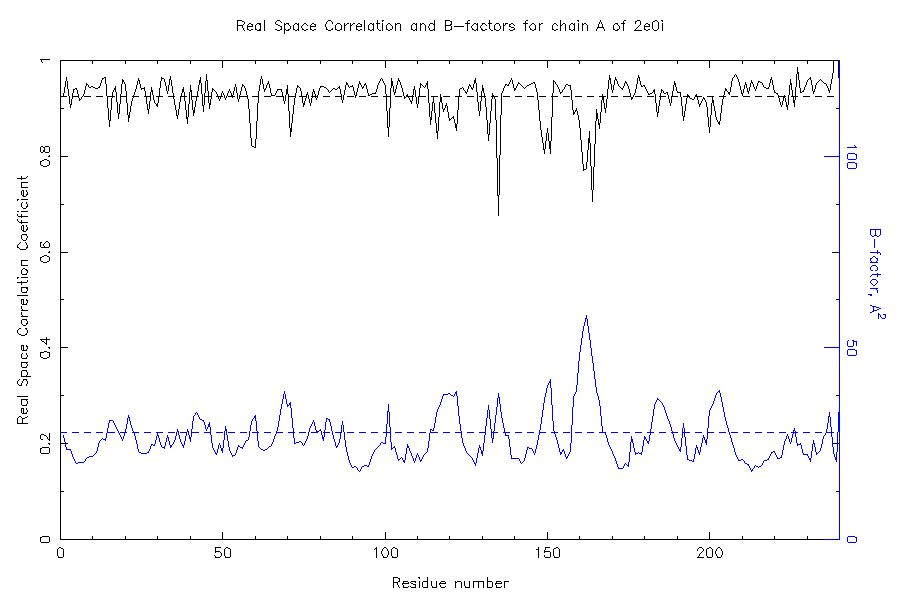
FYI, I attach a real space correlation fit against bias-
minimizing Shake&warp maps of a model I refined at 1.6, where
in final cycles distances and planarities despite
good rsfit for the residues went astray with default weights,
whereas .2 kept things in check for 6 cycles while approaching
stable convergence at rf 20.5 r 17.9. if you overdo it, whatcheck
will tell you that restraints are too tight (which has many
caveats as well as WC does not know about the context of your
structure - floppy vs rigid etc).
I think the plot should convince that at least in this case there
is little to blame on the model quality. I am still trying
more tests, will report if I find anything worthwhile. It is
certainly true that a bad model cannot be saved by tightening
restraints and lower xray weights, but proper adjustment of weights
under consideration of the data resolution and quality as well as
particulars of the model (is it highly flexible, does it need
more restraints etc) while monitoring rfree is a good thing
(and, if you have true atomic resolution, screw what-check).
Till then, best, br
> From: owner-ccp4bb@dl.ac.uk [mailto:owner-ccp4bb@dl.ac.uk]On Behalf Of
> Bart Hazes
> Sent: Saturday, July 14, 2001 8:07 PM
> To: Huiying Li
> Cc: Bernhard Rupp; ccp4bb@dl.ac.uk
> Subject: RE: [ccp4bb]: planarity restraint in refmac5
>
>
> *** For details on how to be removed from this list visit the ***
> *** CCP4 home page http://www.ccp4.ac.uk ***
>
>
> If overall geometric distortions are high then it is likely that the X-ray
> weight was indeed too high. However, local distortions are more
> likely due to
> errors in the model. If an aromatic ring is distorted there is
> not much point
> in flattening it out again. Same for distorted bond lengths and
> angles. Fixing
> these is well within the radius of convergence of the refinement
> program. If
> you see these localized distortions it normally means that
> something else in
> that area is wrong; e.g. VDW conflict, model error... These may well need
> human intervention and should be warning flags to you during the
> next manual
> rebuilding session.
>
> Bart
>
> On Fri, 13 Jul 2001, Huiying Li wrote:
>
> > > My experience in agreement with Garib's advice is that the
> > > X-ray weights are probably set high in default. I occasionally
> > > run a regularization and refine few cycles with x-ray weights
> > > low (Garib suggested 0.1, or lower at low res ( < 3.0)).
> > >
> >
> > Thank you for pointing this out. I re-run the same REFMAC5
> refinement with
> > a weight at 0.1, the distortion in geometry is negligible. The default
> > of X-ray weight in CCP4i (0.5) is apparently way too high for the data
> > resolution (2.1 A) I have.
>
> ==================================================================
> =============
>
> Dept. of Medical Microbiology & Immunology
> University of Alberta
> 1-15 Medical Sciences Building
> Edmonton, Alberta, T6G 2H7, Canada
> phone: 1-780-492-0042
> fax: 1-780-492-7521
>
> ==================================================================
> =============
>
>
2e0i_a_rsfit.gif
{kind=link}