Minimol package.
Minimol is a simple, lightweight package for the storage and manipulation of atomic coordinate models. Unlike MMDB, which is extremely sophisticated, minimol is designed for maximum simplicity, both in terms of the design of the package, and in terms of the user interface (API). As such it should be comparatively simple to use even for programmers with a minimum of C++ experience.
Other design goals include:
- Any object can be assigned or copied with the obvious results.
- Type and const correctness to aid debugging.
- No (visible) memory management.
- Common crystallographic tasks performed with the minimum of instructions.
The simple and lightweight design is based on ideas by Paul Emsley. The user defined properties were inspired by, and a generalisation of the user defined data in MMDB.
The Minimol Hierarchy
The Minimol hierarchy consists of a nest of objects of the following types:
- clipper::MiniMol/clipperMModel. An MModel represents a single coordinate model. Its only contents are a list of MPolymer-s. A MiniMol is an extension of an MModel to include a spacegroup and cell; it can be thought of as an MModel embedded in a crystallographic frame.
- clipper::MPolymer (clipper::MChain). An MPolymer represents a chain of repeating unit (e.g. amino or nucleic acids). It may also be referred to as an MChain, although the generic name is used in this documentation. It consists of a string identifier (see s_mm_polymer_id), and a list of MMonomer-s.
- clipper::MMonomer (clipper::MResidue). An MMonomer represents a single subunit of a repeating sequence (e.g. an individual amino or nucleic acid). It may also be referred to as a MResidue, although the generic name is used in this documentation. It consists of a string identifier (see s_mm_monomer_id), a string describing the monomer type (e.g. LYS, VAL), and a list of MAtom-s.
- clipper::MAtom. An MAtom is an individual atom in the structure. It consists of a string identifier (see s_mm_atom_id). The rest of its properties are inherited from clipper::Atom, from which it is derived.
In addition to the features described above, every Minimol object is a clipper::PropertyManager, which means you can add additional named object of any type to any object. Thus, for example, an atom can also carry a covariance matrix of its coordinates, even though this is not part of the clipper::Atom definition.
- Example hierarchy
- The following is an example of a Minimol hierarchy. Each object may be indexed either by its unique ID, or by an index number. The indices are shown in brackets on the following diagram. Therefore, if the hierarchy is stored in an object called 'minimol', then MPolymer 'A' can be referred to as 'minimol[0]', and MAtom 'A/2/C' can be referred to as 'minimol[0][1][2]'.
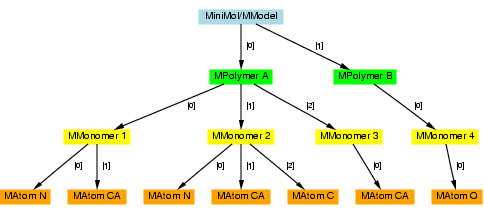
- Comparison to the MMDB hierarchy
- The Minimol hierarchy is similar, but not identical to the MMDB hierarchy. The principle differences are:
- There is no support for multiple models within the hierarchy. If you need more than one model, use more than one object.
- There is no way of navigating up a hierarchy, e.g. from an atom to its residue. This greatly simplifies the package at the cost of some flexibility. One benefit is that there is no distinction between objects which are part of a hierarchy and those which are not.
Common methods among Minimol objects.
Most Minimol objects have common methods wherever possible, so that only one set of function calls need be learned.
The 'child' objects, i.e. clipper::MAtom, clipper::MMonomer, clipper::MPolymer all implement 'id()' and 'set_id()' methods. These methods all the string ID of the atom, monomer, or polymer to be set. These ID's should usually be unique within a particular parent object, although this restriction is not imposed. The format of the IDs is described in s_mm_atom_id, s_mm_monomer_id, s_mm_polymer_id.
The 'parent' objects, i.e. clipper::MMonomer, clipper::MPolymer, clipper::MModel (and therefore clipper::MiniMol) have the following common methods:
- size() returns the number of children of this object, e.g. the number of MAtoms in an MMonomer.
- [int] array subscription operators return the child specified by the index, in the range 0...size()-1
- find(string) associative access operators return the child with the specified ID. If no such child exists, an error occurs (See lookup).
- select(string) returns a copy of this hierarchy containing only those objects specified by the selection string.
- transform(RTop_orth) applies a coordinate transformation to an entire sub-hierarchy.
- atom_list() returns a clipper::Atom_list containing all the atoms in the subhierarchy of this object.
In addition to these methods, hierarchies can be combined using the logical '&' and '|' operators; the former returns those objects common to both hierarchies, and the latter returns the union of the two hierarchies.
Assigning and copying Minimol objects.
All Minimol objects may be assigned, copied, and passed to subroutines with no harmful side effects. Passing a Minimol object creates a copy of that object, all of its properties, and all of its children (i.e. the whole hierarchy under the object).
Assigning a Minimol object destroys whatever was previously in the destination object, replacing it with a copy of the source object, as above.
If you do not wish to make a copy of hierarchy or portion of a hierarchy, the copy() method of each object allows fine grained control over what is copied.
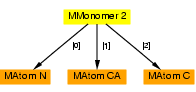
For example, if using the hierarchy from s_mm_hierarchy we were to give the assignment command:
clipper::MMonomer monomer = minimol[0][1];
Then 'monomer' would contain the following sub-hierarchy:
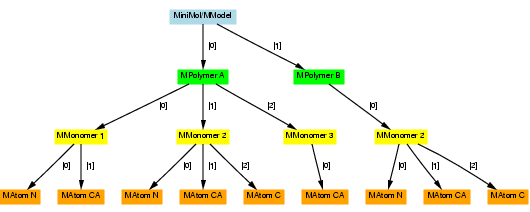
As well as using assignments to copy sub-hierarchies out of a hierarchy, we can use them to duplicate hierarchies or copy a sub-hierarchy back into a hierarchy. For example, following the previous code, we could give the following assignment:
minimol[1][0] = monomer;
The data from 'monomer' and its children then replace the original monomer at minimol[1][0], giving the following hierarchy:
Selection and logical functions
Selections can be applied at any level of the hierarchy using the 'select()' method of the top object in the hierarchy. The selection string consists of a comma separated list of allowed ID's for each level of the hierarchy below the current, with the levels separated by slashes '/'. The asterisk character '*' selects all object on a level.
e.g. the selection string "* /13,14,15/CA" applied at the model level selects the C-alpha atoms of residues 13, 14 and 15 of every chain. For details of the individual IDs, see the following three section.
The select functions always return a complete hierarchy starting with the current object, containing all the specified child objects. Select functions are commonly used in combination with logical operators, to combine the results of several selections. The logical '&' and '|' operators are provided; the former returns those objects common to both hierarchies, and the latter returns the union of the two hierarchies.
Polymer IDs.
Polymer IDs may be any string, however for PDB-derived hierarchies it is traditional for the polymer ID to be a single upper-case letter.
Monomer IDs.
Monomer IDs are based on a sequence number and optionally an insertion code. The sequence number is the numeric position of the monomer within the sequence, although this is not a rigid convention. In practise any numbering convenient to the problem at hand is used. In some cases, extra residues are inserted into a sequence: These are commonly represented by an insertion code appended to the sequence number. The insertion code is commonly a single upper-case letter.
Monomer IDs in Minimol are formatted as a 4-character string containing the right-justified sequence number. (Larger numbers cause this field to be expanded). If an insertion code is present, then a colon ':' and the insertion code are appended to the end of the ID. e.g. "___1", "_123", "_123:B".
When a monomer ID is supplied to Minimol as part of a select() or find() method, the insertion code is removed, the numeric part is right justified and the insertion code reapplied. Therefore in all common cases the string representation of the sequence number may be used to refer to a monomer within a polymer. The same transformation is applied when the 'set_id()' function is used.
Atom IDs.
Atom IDs are based on the element of the atom, and on the position of the atom within the monomer.
Atom IDs in Minimol follow the PDB convention of a 4-character string. The first 2 characters are the right-justified upper case element name. The next 2 characters (usually a letter and optional number) refer to the position within the monomer. e.g. "_CA_", "_N__", "_NZ1", "ZN__".
If multiple conformations are present, a colon ':' and the alternate conformation code (usually a single uppercase letter) are appended to the end, e.g. "_NZ1:A".
When an atom ID is supplied to Minimol as part of a select() or find() function, the length of the ID is checked. If there are 4 characters in total, or 4 characters before a colon, then the ID is left as it is. Otherwise the portion before the colon (if any) is justified assuming a single character atom name, unless the second character is lower case, in which case a two character atom name is assumed and converted to upper case. Therefore in the common cases an unjustified string representation of the atom ID may be used to refer to an atom within a monomer.
Reading and writing to MMDB or file.
Minimol provides a convenient mechanism for communicating coordinate models to and from MMDB, and therefore to or from PDB and CIF files, through the clipper::MMDBfile class. This object is a trivial derivation of the clipper::MMDBManager and CMMDBManager objects, so that reference to such objects can be safely cast to a clipper::MMDBfile. Alternatively, for file access, the clipper::MMDBfile class can be used directly.
The clipper::MMDBfile class provides convenient read_file() and write_file() method. These provide no additional functionality over the underlying MMDB methods, but can be called with standards strings for the filenames.
The clipper::MMDBfile class also provide two additional methods which allow a MiniMol object to be imported from, or exported to an MMDB.
- The clipper::MMDBfile::import_minimol() method imports either the whole of an MMDB hierarchy, or an MMDB selection from that hierarchy into MiniMol. In addition to the normal atom, residue and chain data, each object is labelled with an optional property, "CID", which contains the MMDB ID describing where in the MMDB hierarchy the object came from. This information may optionally be used to later restore information from Minimol back into the same objects in the MMDB hierarchy.
- The clipper::MMDBfile::export_minimol() method exports information from a MiniMol back into MMDB. If the Minimol objects have "CID" properties and those CIDs exist in the MMDB hierarchy, then the information in the Minimol objects is used to update the corresponding objects in the MMDB hierarchy. If there are either no "CID" properties, or the CIDs don't exist in the MMDB hierarchy, new object are created in MMDB using the information from Minimol. Thus the export function may be used either to fill an entire MMDB from a MiniMol, or to update MMDB objects which have been modified in Minimol. However mixing these two approaches in a single export can lead to confusing results!
For examples of communication between MiniMol and MMDB, see the following section.
Minimol Examples.
To import a MiniMol from an existing MMDB, the following code can be used:
CMMDBManager mmdb; // Initialise MMDB here clipper::MiniMol mmol; static_cast<clipper::MMDBfile&>(mmdb).import_minimol( mmol );
To import a MiniMol from a file, the following code can be used:
clipper::MMDBfile mfile; clipper::MiniMol mmol; mfile.read_file( "input.pdb" ); mfile.import_minimol( mmol );
To export an entire MiniMol to a file, the following code can be used:
clipper::MMDBfile mfile; mfile.export_minimol( mmol ); mfile.write_file( "output.pdb" );
To print the polymer IDs, monomer IDs and atom IDs of every atom in a MiniMol, use the following:
for ( int p = 0; p < mol.size(); p++ ) for ( int m = 0; m < mol[p].size(); m++ ) for ( int a = 0; a < mol[p][m].size(); a++ ) std::cout << mol[p].id()+"\t"+mol[p][m].id()+"\t"+mol[p][m][a].id()+"\n";
The following subroutine mutates a residue from one type to another, by finding the operator which maps the reference residue onto the target residue, overwriting the target residue with the reference residue (apart from the ID, which is preserved), and then transforming the new residue into place.
void mutate_residue( MMonomer& from, const MMonomer& to ) { Atom_list frlist, tolist; // make lists of cardinal atoms frlist.push_back( from.find( "CA", MM::ANY ) ); // get old atoms frlist.push_back( from.find( "C", MM::ANY ) ); frlist.push_back( from.find( "N", MM::ANY ) ); tolist.push_back( to.find( "CA", MM::ANY ) ); // get new atoms tolist.push_back( to.find( "C", MM::ANY ) ); tolist.push_back( to.find( "N", MM::ANY ) ); RTop_orth rtop( tolist, frlist ); // calc transform String id = from.id(); // save old ID from = to; // copy in new residue from.set_id( id ); // with old ID from.transform( rtop ); // and old position }
