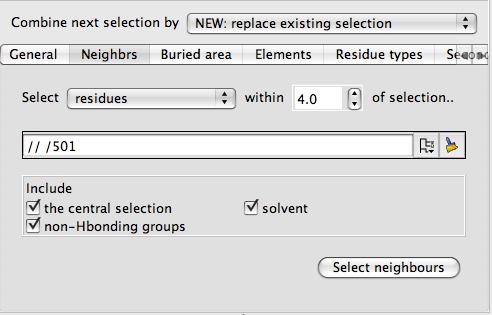
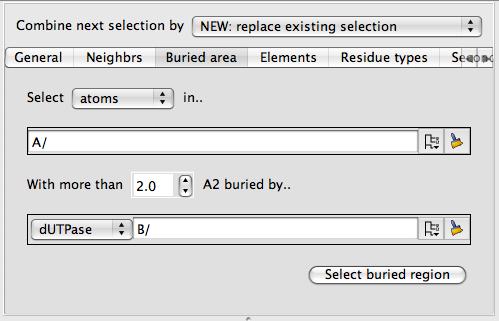
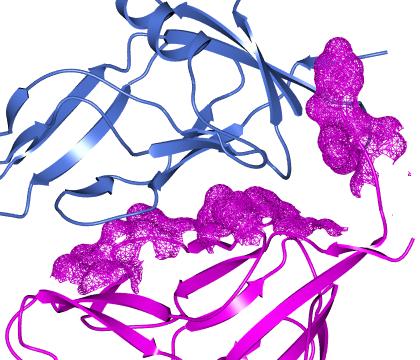
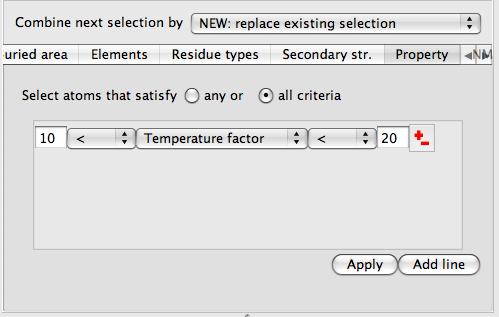
As in the example above you should select two groups of atoms (such as two different chains) and click Select buried region. Beware that this tool may be computationally expensive. If you are applying this tool to a surface then you will also need to select the context set of atoms (the second column in the display table)
which will usually be the same as the first set of atoms in the buried area tool.
The residue name and atom element type are usually redundant, that is they are not essential to uniquely identify an atom, and do not need to be included when entering a CID. But note that all spaces in the coordinate ID are ignored and this means that certain
atom names will be interpreted correctly only if the chemical element
name is supplied (compare Calcium CA[CA] and
Carbon in alpha-position CA[C]).
A coordinate ID may be incomplete. Below are the rules for
interpretation of incomplete IDs. Curling brackets
{..} denote parts of an ID string that may
be omitted as a whole:
| Coordinate ID |
Description |
| /1/A/33(SER).B/CA[C].A |
model 1, chain A, residue SER with sequence
number 33 and insertion code B, C-alpha atom in alternative
location A. |
| /1/A/*(SER).* |
any SER residue in chain A, model 1.
|
| /1/A/(SER) |
any SER residue with no insertion code in chain A, model 1. |
| /1//(SER) |
any SER residue with no insertion code in chain without a chain ID, model 1. |
| /1/A/*.*/CA[C] |
any C-alpha atom with no alternative location indicator in chain A, model 1, in residues with any sequence number and insertion code. |
| /1/A/*/CA[C] |
any C-alpha atom with no alternative location indicator in chain A, model 1, in residues with any sequence number and no insertion code. |
| A/*/CA |
any C-alpha or Calcium atom with no alternative location indicator in chain A of any model, in residues with any sequence number and no insertion code. |
| CA[C] |
any C-alpha atom with no alternative location indicator in any chain, any model, in residues with any sequence number and no insertion code. |
| CA |
any atom of chain CA with no alternative location indicator, in any model, in residues with any sequence number and no insertion code. |
Selection aliases are one word aliases for complex CIDs. The predefined selection aliases are specified in the file ccp4mg/python/ui/selection_protocols.py. This needs a nice GUI to allow customisation. Within the program the implementation of selection aliases is different from selection commands but the user need not be concerned by this.
| amino_acid |
All amino acid residues - defined as a list of residue types |
| nucleic |
All nucleic acid residues - defined as a list of residue types |
| solvent |
All solvent - defined as a list of residue types |
| solute |
All solute - defined as a list of residue types |
| catrace |
All atoms called 'CA' in amino acid residues |
| peptide_link |
The main chain atoms N,C,O,H |
| main |
All main chain atoms in amino acid residues |
| side |
All not main chain atoms in amino acid residues |
| ca+side |
All CA and side chain atoms in amino acid residues |
| backbone |
Atoms C3*,O3*,H3*,C4*,C5*,1H5*,O5*,2H5*,H5T,P,O1P,O2P |
| sugar_ring |
Atoms C1*,H1*,C2*,2H2*,1H2*,C3*,O3*,H3*,C4*,H4*,C5*,1H5*,2H5*,O5*,H5T,O4*,O2* |
| base |
Defined as the atoms which are not backbone or sugar_ring |
| polar_atoms |
Defined as the atoms of element type N,O,P |
More complex selections can be defined by a selection command which consists of a command keyword and a variable number of arguments.
The format for commands is:
command [arg1 [arg2 .... [argn]]]
with the arguments in the order given in the descriptions below.
Currently supported commands and their arguments:
| neighb |
Select atoms, residues or chains within neighbourhood of given object |
| cid |
An ID for the central atom, residue or chain |
| maxd |
The maximum distance from the central atom to the neighbours |
| group |
Select all atoms in a group that have at least one atom within maxd distance. The currently supported groups are: model,chain,residue,main,side,catrace,main_side,CA+side,solvent_monomers. |
| hbonded |
If this has value 1 then only hydrogen bonded atoms are selected. |
| excl |
A comma separated list of atoms which will be excluded from the selection: central (the 'central' atoms defined by cid argument), solvent, monomer (all monomers), peptide (main chain atoms excluding CA). |
| sphere |
Select atoms, residues or chains within radius of a given x,y,z coordinate |
| x |
X coordinate |
| y |
Y coordinate |
| z |
Z coordinate |
| radius |
The maximum distance from the central point |
| type |
Should be 'atom','residue' or 'chain' to specify type of objects selected |
| all |
|
>Select all atoms in the model |
| oneatom |
|
>Select one atom per residue in the model |
| termini |
|
Select the termini residues of chains |
| symid |
Select the model containing coordinates for the symmetry mate |
| n_xyz |
symmetry id |
Complex selection commands are built up from CIDs, aliases and commands connected by the operators
The selection components can also be grouped by using curly brace. Note that curly brace are used to be distinct from the brackets used to denote residue types in the CID.

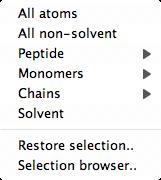 The options on the atom selection menu are customised for the data; for example
the chains and monomers in the structure will be listed where appropriate.
Several options on the selection menu have pull-right sub-menus for
selecting individual items (for example for Monomers).
The options on the atom selection menu are customised for the data; for example
the chains and monomers in the structure will be listed where appropriate.
Several options on the selection menu have pull-right sub-menus for
selecting individual items (for example for Monomers).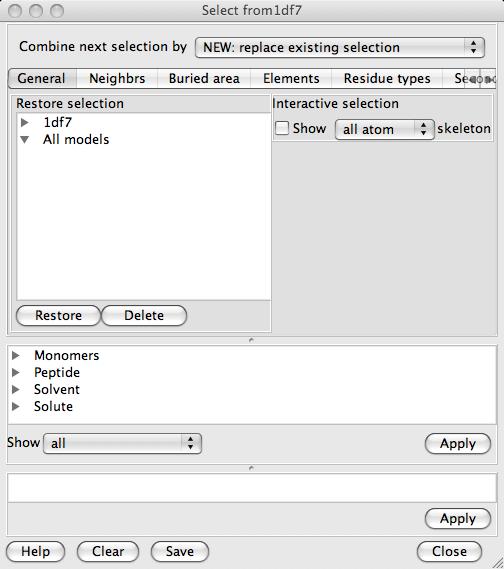 The browser consists of three vertical sections with three different approaches to atom selection:
The browser consists of three vertical sections with three different approaches to atom selection: